- Exploratory Analysis
Petr V. Nazarov, LIH
2018-03-05
3.1. Check data distribution
rm(list=ls()) ## clean memory
## download the data (in case you have not downloaded it before)
setwd("D:/Data/R") # sets the current folder
download.file("http://edu.sablab.net/rt2018/data/LUSC60.RData", destfile="LUSC60.RData",mode = "wb")Now let’s investigate the data: see the content and distributions (remember - right skewed data are hard to work with!)
load("LUSC60.RData") # load the data
ls()## [1] "Anno" "color" "Data" "ensg" "LUSC" "oncogenes"
## [7] "X" "XAnno"## what is inside?
str(LUSC)## List of 5
## $ nsamples : num 60
## $ nfeatures: int 20531
## $ anno :'data.frame': 20531 obs. of 2 variables:
## ..$ gene : chr [1:20531] "?|100130426" "?|100133144" "?|100134869" "?|10357" ...
## ..$ location: chr [1:20531] "chr9:79791679-79792806:+" "chr15:82647286-82708202:+;chr15:83023773-83084727:+" "chr15:82647286-82707815:+;chr15:83023773-83084340:+" "chr20:56064083-56063450:-" ...
## $ meta :'data.frame': 60 obs. of 5 variables:
## ..$ id : chr [1:60] "NT.1" "NT.2" "NT.3" "NT.4" ...
## ..$ disease : chr [1:60] "LUSC" "LUSC" "LUSC" "LUSC" ...
## ..$ sample_type: chr [1:60] "NT" "NT" "NT" "NT" ...
## ..$ platform : chr [1:60] "ILLUMINA" "ILLUMINA" "ILLUMINA" "ILLUMINA" ...
## ..$ aliquot_id : chr [1:60] "a7309f60-5fb7-4cd9-90e8-54d850b4b241" "cabf7b00-4cb0-4873-a632-a5ffc72df2c8" "ee465ac7-c842-4337-8bd4-58fb49fce9bb" "6178dbb9-01d2-4ba3-8315-f2add186879a" ...
## $ counts : num [1:20531, 1:60] 0 3 13 272 1516 ...
## ..- attr(*, "dimnames")=List of 2
## .. ..$ : chr [1:20531] "?|100130426" "?|100133144" "?|100134869" "?|10357" ...
## .. ..$ : chr [1:60] "NT.1" "NT.2" "NT.3" "NT.4" ...plot(density(LUSC$counts),lwd=2,col="blue",main="Data distribution")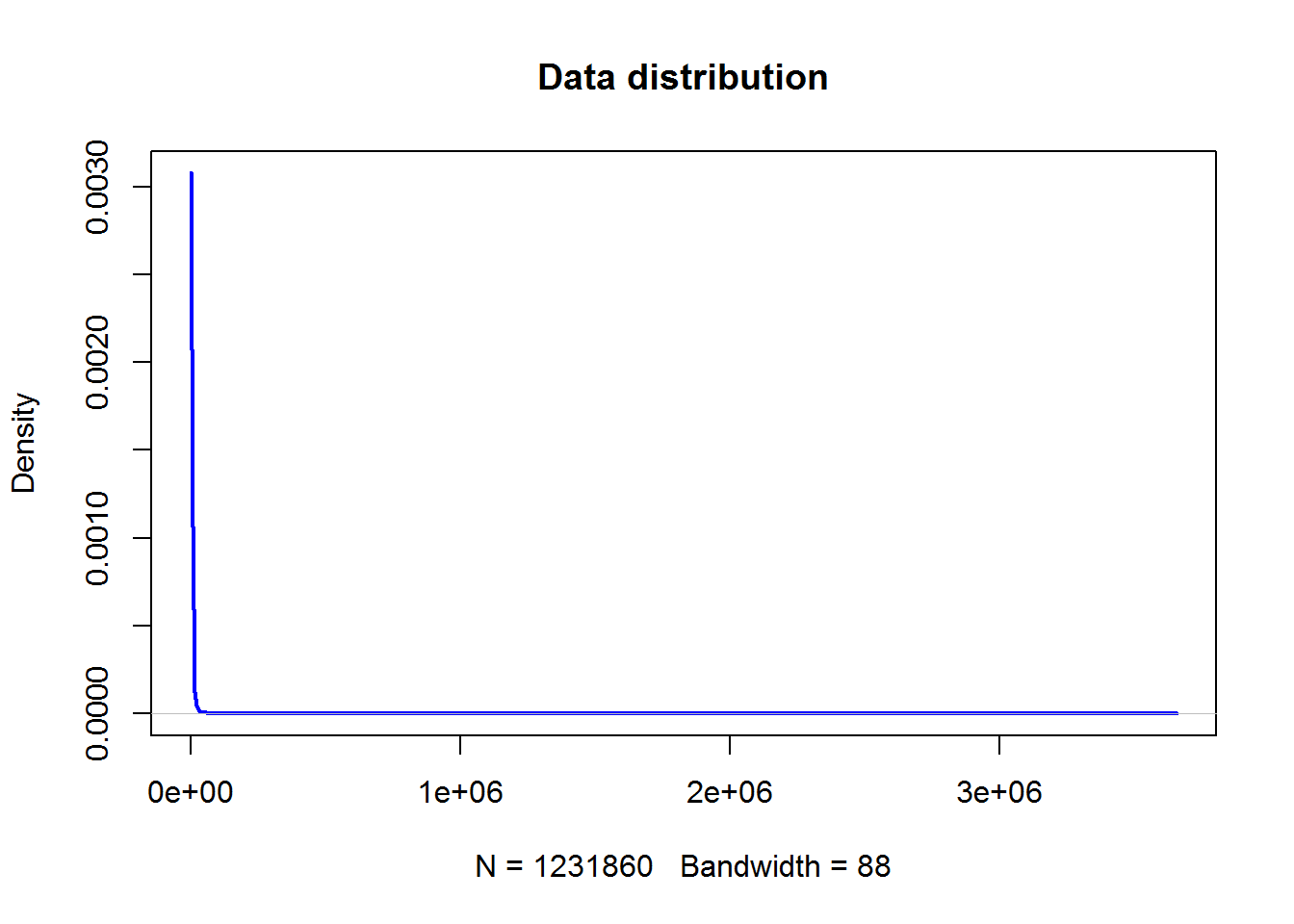
Higly right skewed! Need to do log2 transformation!
X = log2(1+LUSC$counts)
plot(density(X),lwd=2,col="blue",main="Data distribution (log scale)")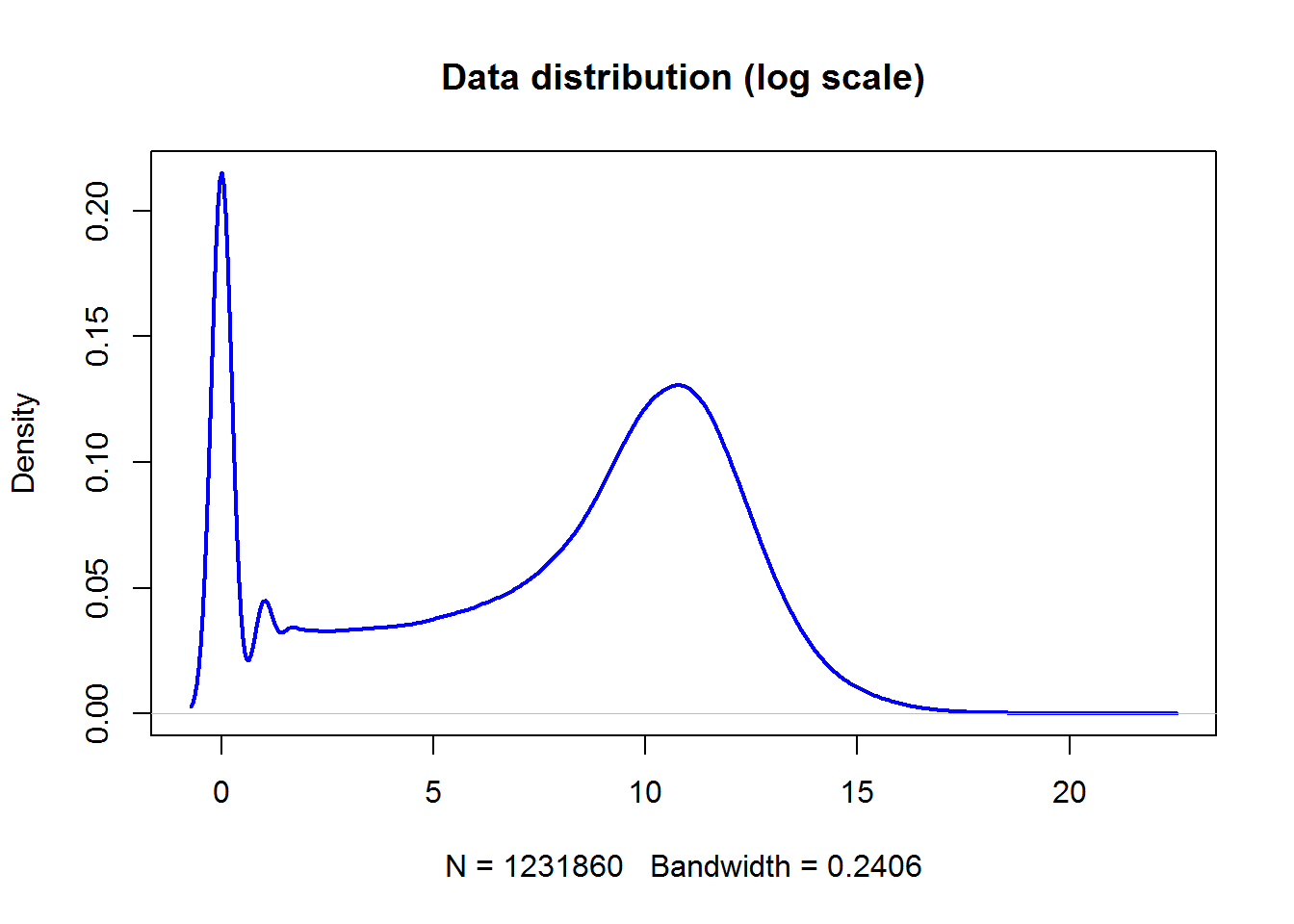
You can use my warp-up function plotDataPDF to see distribution of each samples
## load the script / function
source("http://sablab.net/scripts/plotDataPDF.r")
## define colours
summary(as.factor(LUSC$meta$sample_type))## NT TP
## 20 40color = c(TP = "#FF000055", NT = "#0000FF55")[LUSC$meta$sample_type]
## use warp-up function
plotDataPDF(X,lwd=2,col=color,main="Distributions of gene expression in each sample")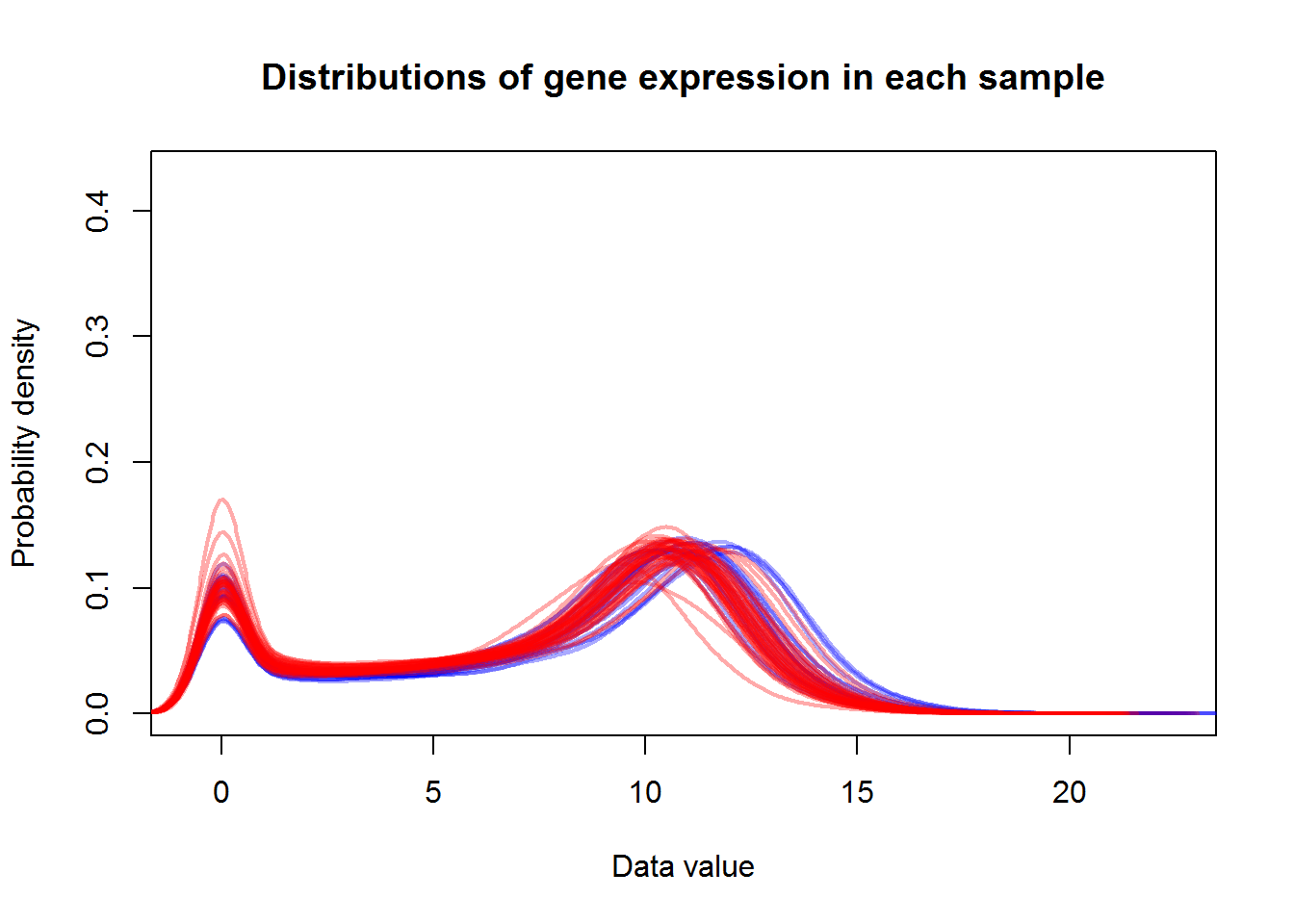
Conclusion: Data were highly right skewed. They were log2-transformed. Distributions are comparable.
3.2. PCA - Prinicipal Component Analysis
The standard function for PCA in R is prcomp. It considers rows as objects and columns as features. There are several limitations of prcomp:
It works only with
matrixobjects. => Please, transform yourdata.frametomatrixusingas.matrix()function.No NA values are allowed (please, impute or remove the features containing them)
No features of 0 variance are allowed (please, remove features that all are zeros)
There are more advanced PCA tools - Tony could comment :)
## perform PCA
PC = prcomp(t(X))
str(PC)## List of 5
## $ sdev : num [1:60] 119.2 72.6 46 39.3 36 ...
## $ rotation: num [1:20531, 1:60] 1.65e-15 6.69e-05 -8.65e-04 7.38e-04 1.24e-03 ...
## ..- attr(*, "dimnames")=List of 2
## .. ..$ : chr [1:20531] "?|100130426" "?|100133144" "?|100134869" "?|10357" ...
## .. ..$ : chr [1:60] "PC1" "PC2" "PC3" "PC4" ...
## $ center : Named num [1:20531] 0 4.85 4.8 8.6 11.1 ...
## ..- attr(*, "names")= chr [1:20531] "?|100130426" "?|100133144" "?|100134869" "?|10357" ...
## $ scale : logi FALSE
## $ x : num [1:60, 1:60] -89.7 -128.5 -100.9 -159.4 -196.4 ...
## ..- attr(*, "dimnames")=List of 2
## .. ..$ : chr [1:60] "NT.1" "NT.2" "NT.3" "NT.4" ...
## .. ..$ : chr [1:60] "PC1" "PC2" "PC3" "PC4" ...
## - attr(*, "class")= chr "prcomp"## plot PC1 and PC2 only
plot(PC$x[,1],PC$x[,2], col=color, pch=19)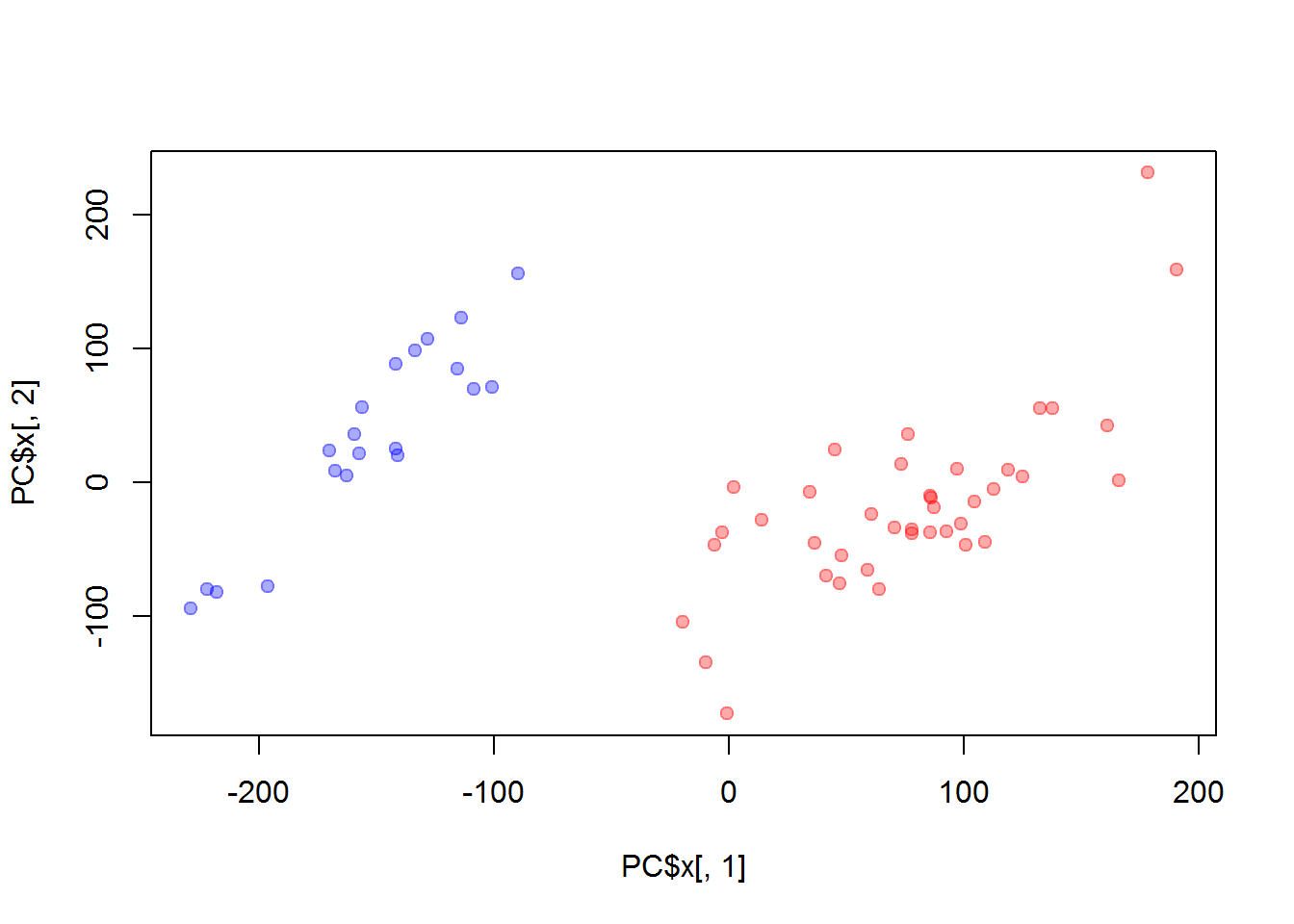
If you want, you can use a warp-up function for PCA plotPCA(). It takes care about some limitations (NAs, zero variance). No need to transpose data.
## load the script
source("http://sablab.net/scripts/plotPCA.r")
## use our warp-up for PCA
plotPCA(X, col=color, pch=19, cex=2, cex.names=0.5)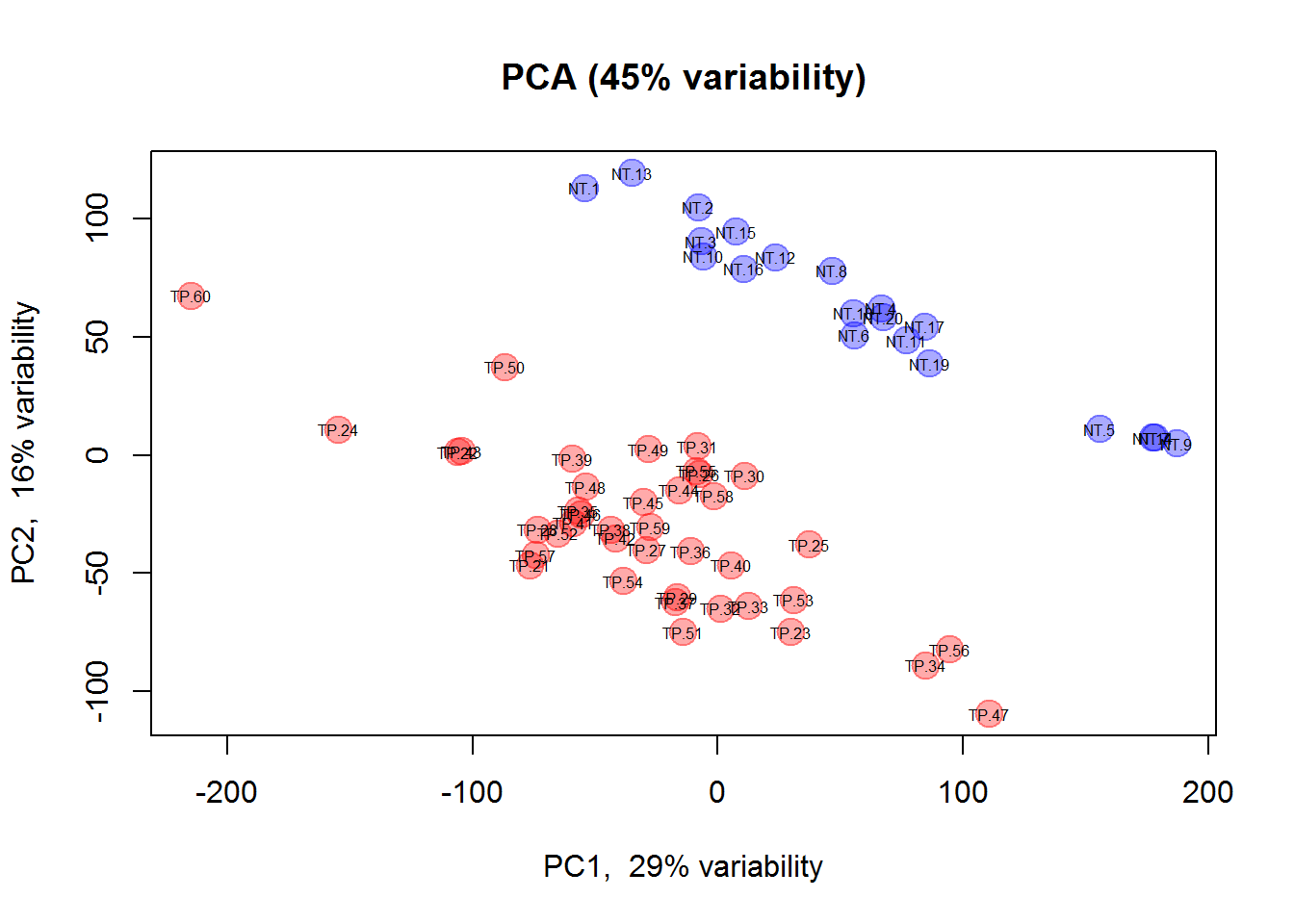
## add parameter scale=FALSE to get the same picture as beforeYou can also use 3D visualization:
## install package for 3D visualization
install.packages("rgl")
## load the package
library(rgl)
plot3d(PC$x[,1],PC$x[,2],PC$x[,3],
size = 2,
col = color,
type = "s",
xlab = "PC1",
ylab = "PC2",
zlab = "PC3")We considered samples as objects and genes as features. Now let’s do vice versa: genes as objects and samples as features.
## perform PCA - no transposition here
PC2 = prcomp(X)
## plot PC1 and PC2 only
plot(PC2$x[,1],PC2$x[,2], col="#00880055", pch=19, cex = apply(X,1,mean)/10)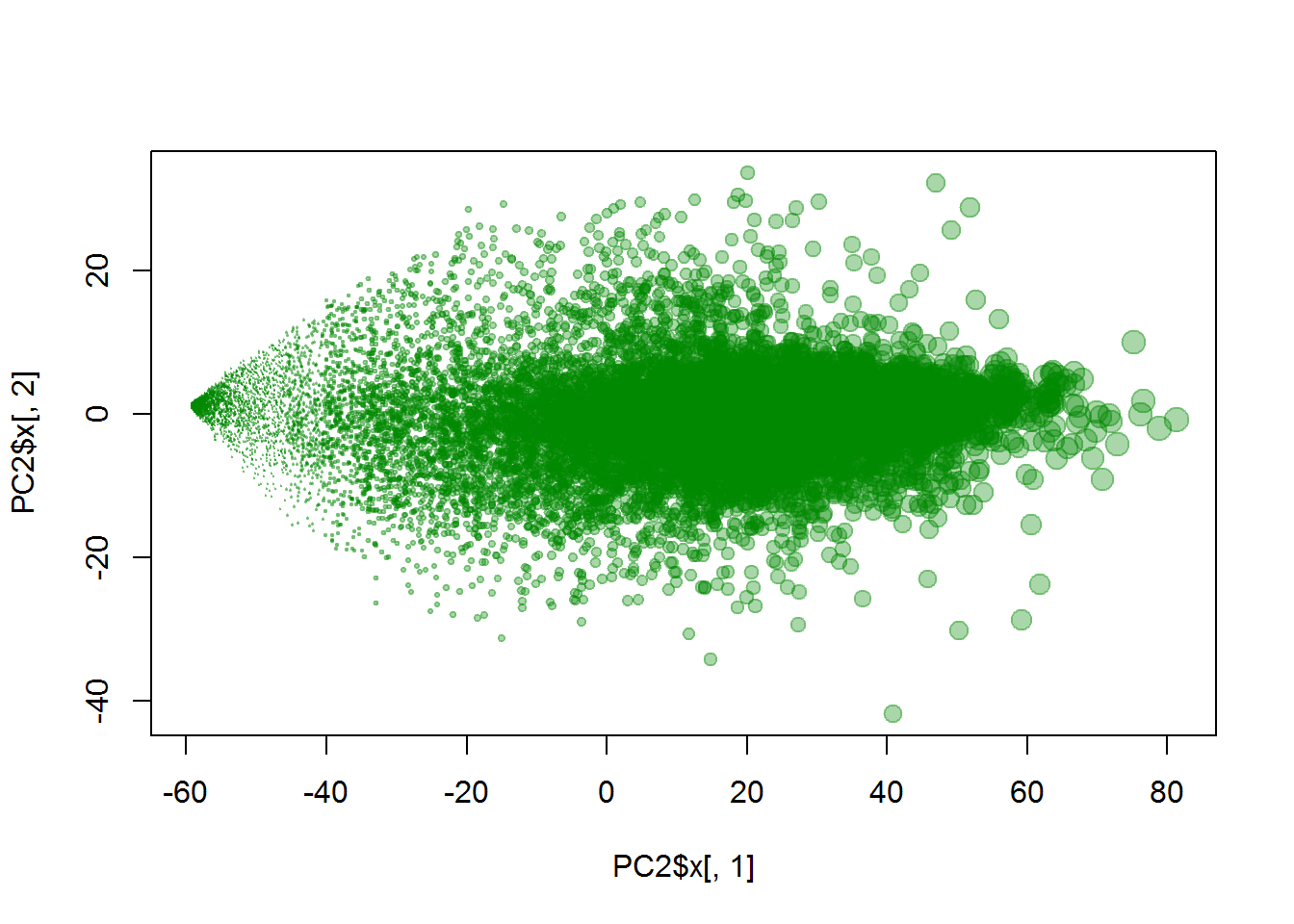
## for fun we used average expression to assign dot size :)
3.3 Correlations
Correlation can show linear agreement b/w samples (through the genes) or genes (through the samples). Let’s start with the first one. Please consider the range of correlation - this is usially the case for sample correlation in RNA-seq and one-colour arrays. Usually people use either Pearson (method="pearson") or Spearman (method="spearman").
RS = cor(X) ## try adding parameter method="spearman"
str(RS)## num [1:60, 1:60] 1 0.964 0.954 0.958 0.95 ...
## - attr(*, "dimnames")=List of 2
## ..$ : chr [1:60] "NT.1" "NT.2" "NT.3" "NT.4" ...
## ..$ : chr [1:60] "NT.1" "NT.2" "NT.3" "NT.4" ...## plot correlation matrix as a heatmap
library(pheatmap)
plot(density(RS),lwd=2,col="blue",main="Distribution of sample-sample correlation")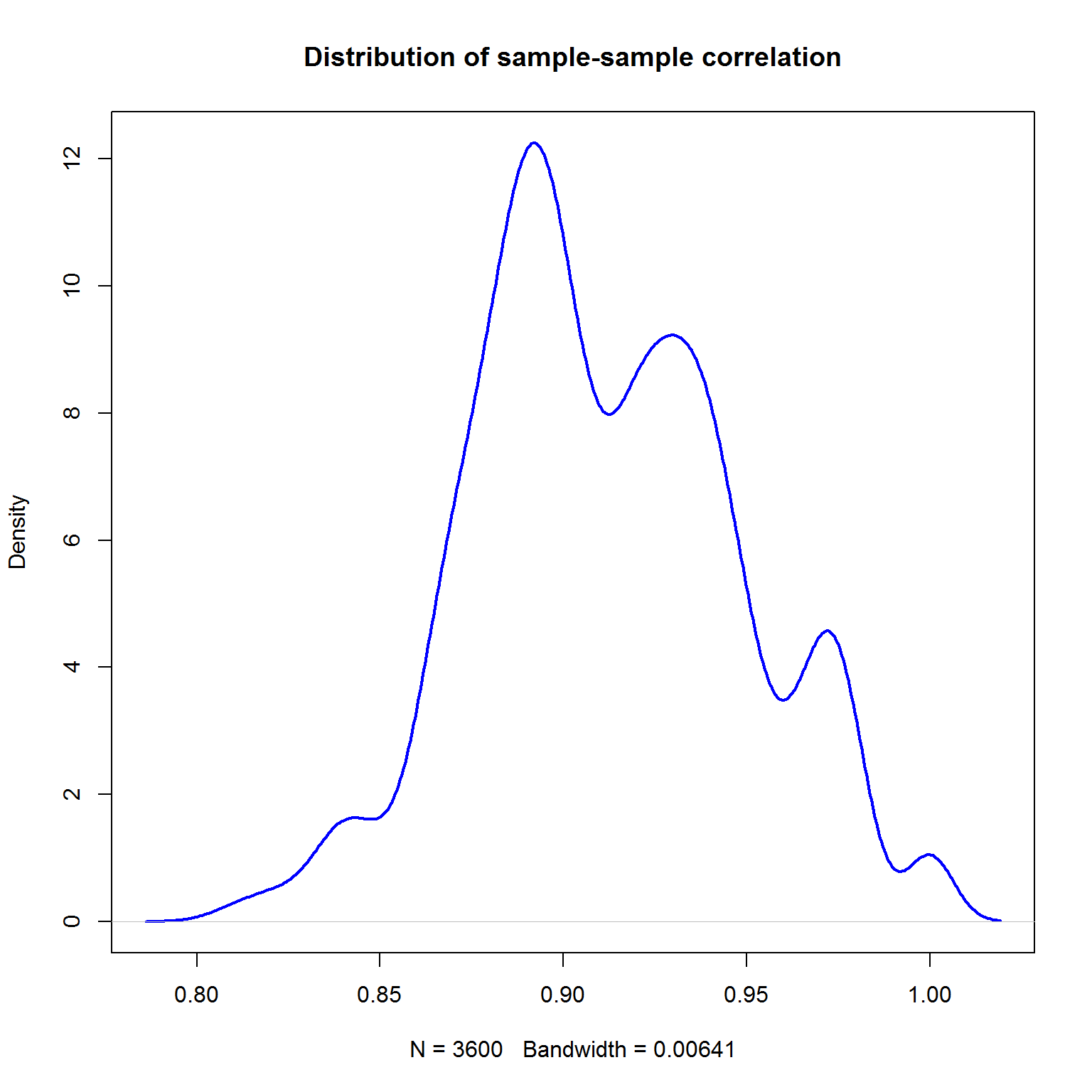
pheatmap(RS,main="Correlation b/w Samples")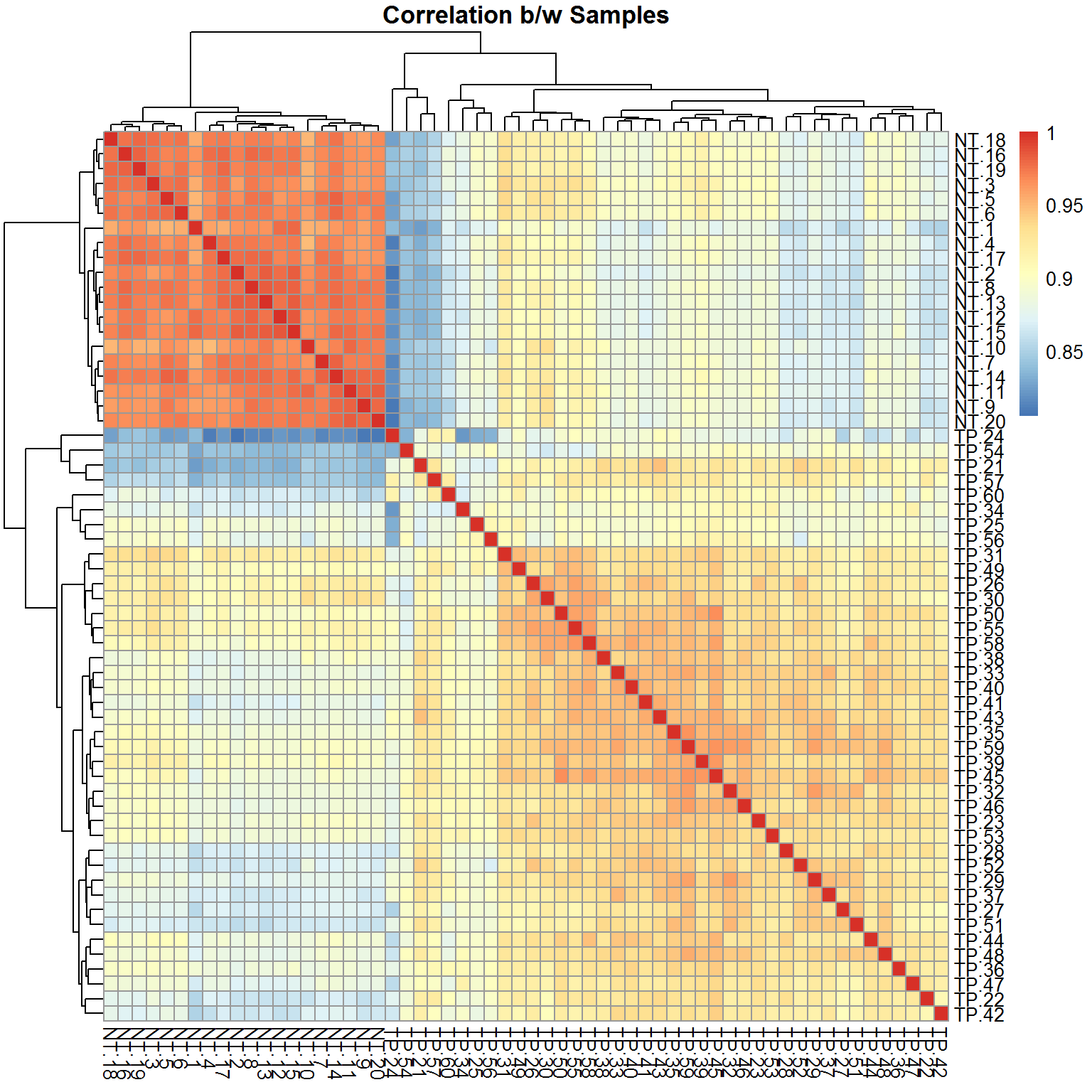
You could also correlate genes. Be prepared to get huge matrix! So, if you can, filter the genes first.
## filter the genes
ikeep = apply(X,1,max) > 7
ikeep = ikeep & apply(X,1,sd) > 1
sum(ikeep)## [1] 9229RG = cor(t(X[ikeep,]))
str(RG)## num [1:9229, 1:9229] 1 0.65 0.664 0.192 0.223 ...
## - attr(*, "dimnames")=List of 2
## ..$ : chr [1:9229] "?|100133144" "?|100134869" "?|155060" "?|340602" ...
## ..$ : chr [1:9229] "?|100133144" "?|100134869" "?|155060" "?|340602" ...plot(density(RG),lwd=2,col="red",main="Distribution of gene-gene correlation")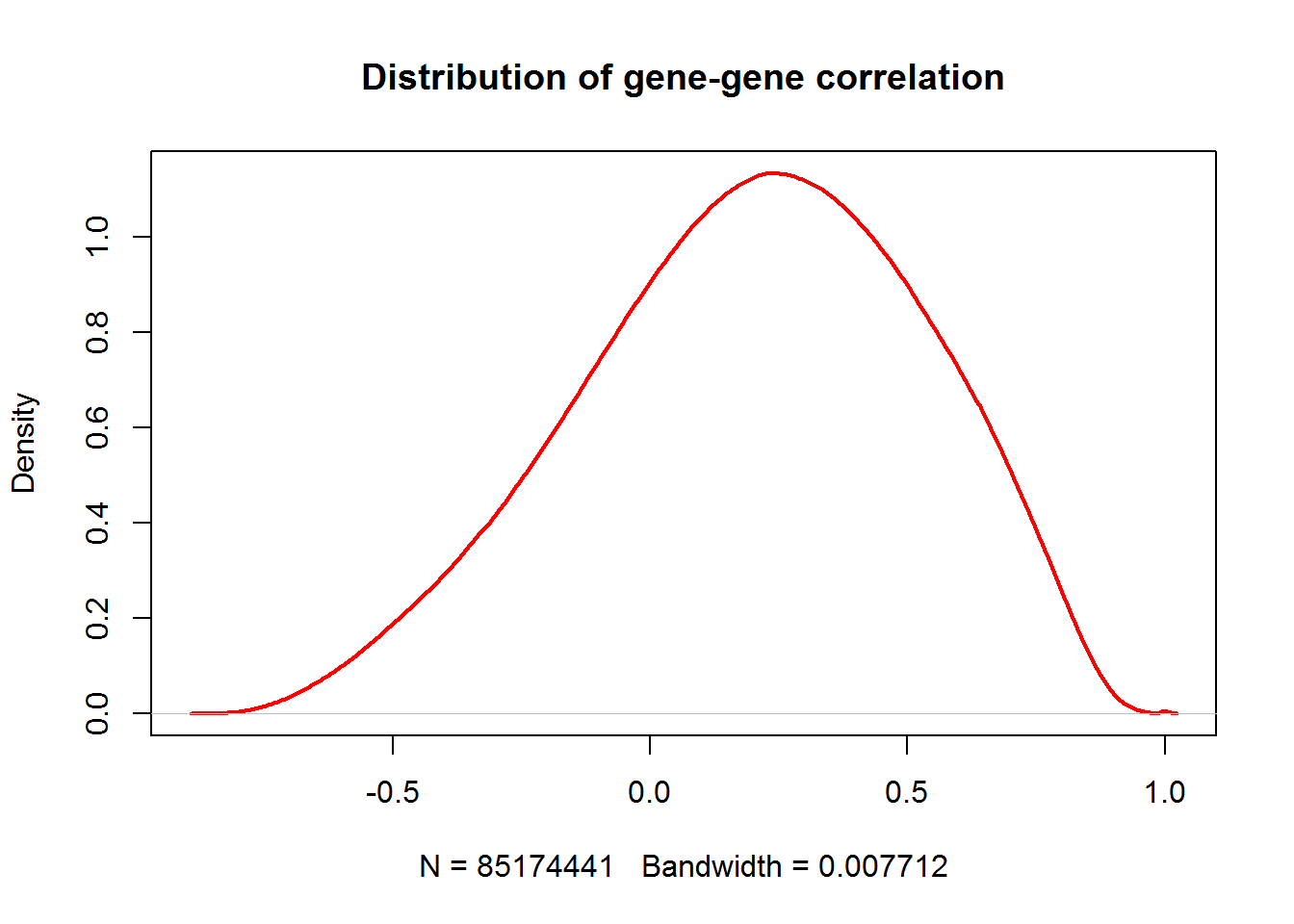
Excercises
- Work with counts.txt data. Build distributions and perform necessary transformations. Perform PCA. Hint: Consider filtering out lowly expressed and invariable genes
density,log,apply,mean,max,sd,prcomp
- Work with HTA.txt data. This dataset was obtained by HTA 2.0 arrays on the same samples as RNA-seq presented in
counts.txt. Build distributions and transform data if needed. Perform PCA.
density,prcomp
![]()